
Warning
This documentation is for CPAT v3.0.0 or future versions. For documentation of CPAT v2.0.0 and older versions, please go to http://rna-cpat.sourceforge.net/
Release history
CPAT v3.0.5 (01/24/2024)
Use “pyproject.toml” to replace “setup.py”.
Note
cpat.pyis now renamed tocpat. Similarly,make_hexamer_tab.pyis renamed tomake_hexamer_tab,make_logitModel.pyis renamed tomake_logitModel.To ensure compatibility with the previous pipelines, please add the following lines into your ~/.bashrc file.
alias cpat.py='cpat'
alias make_hexamer_tab.py='make_hexamer_tab'
alias make_logitModel.py='make_logitModel'
CPAT v3.0.4 (05/26/2021)
Fix bug to read remote file for Python3.
CPAT v3.0.3 (03/08/2021)
Update “cpat.py” to handle alternative start codens.
CPAT v3.0.2 (08/17/2020)
Update “make_logitModel.py” to make it compatible with “cpat.py”.
CPAT v3.0.1
Minor bug fixed regarding the output format.
CPAT v3.0.0
For many transcripts, the longest ORF may not be the real ORF. For example, in human genome, the 2nd longest ORF of NM_198086 is the real ORF, and the 3rd longest ORF of NM_030915 is the real ORF. Version 3.0.0 is released to address this problem.
If model is provided, CPAT can be used as an ORFfinder. It gives exactly the same results as NCBI ORFfinder does.
Search for all ORF candidates. The number of ORF reported is controlled by
--min-orfand--top-orf.In addition to basic ORF information (“ORF frame”, “ORF strand”, “ORF start”, “ORF end”, “ORF sequence”), it also reports “coding probability” for each ORF.
The best ORF will be selected (controlled by
--best-orf) either by ORF length or coding probability.
Introduction
CPAT is a bioinformatics tool to predict RNA’s coding probability based on the RNA sequence characteristics. To achieve this goal, CPAT calculates scores of these 4 linguistic features from a set of known protein-coding genes and another set of non-coding genes.
ORF size
ORF coverage
CPAT will then builds a logistic regression model using these 4 features as predictor variables and the “protein-coding status” as the response variable. After evaluating the performance and determining the probability cutoff, the model can be used to predict the coding potential of new RNA sequences.
Installation
Prerequisite
install CPAT using pip3
$ pip3 install CPAT
$ pip3 install CPAT --upgrade # if you already have CPAT v2.0 installed
Note
User need to download prebuilt logit model and hexamer table for human, mouse, zebrafish and fly. For other species, we provide scripts to build these models (see below).
Run CPAT online
https://wlcb.oit.uci.edu/cpat is hosted by Dr Wei Li’s lab @ University of California Irvine.
Step1: Upload data to CPAT server. There are 3 different ways to uploada
Upload BED or FASTA format files from local disk. Files can be regular or compressed (.gz, .Z. .z, .bz, .bz2, .bzip2).
For small dataset, user can copy and paste data (in BED or FASTA format) directly to the text area.
For larger dataset, user can save data in web server (http, https or ftp) first, then paste the data url to text area. For very large dataset, run CPAT locally.
Step2: Select Select Species assembly
Step3: Click Submit button
Note
This web server only supports Human (hg19), Mouse (mm9 and mm10), Fly (dm3) and Zebrafish (Zv9).
When input file is BED format, the reference genome is required and the assembly version is important.
When input file is FASTA format, the reference genome and the assembly version is ignored.
Run CPAT on local computer
Input files
User needs to provide a gene file (‘-g’), a logit model file (‘-d’), a hexamer frequency table file (‘-x’) and specify the output file name(‘-o’). Gene file could be either in BED or FASTA format. If in BED format, user also needs to specify the reference genome sequence file (‘-r’).
Command line options
$ cpat -h
cpat [options]
Options:
--version show program's version number and exit
-h, --help show this help message and exit
-g GENE_FILE, --gene=GENE_FILE
Genomic sequnence(s) of RNA in FASTA
(https://en.wikipedia.org/wiki/FASTA_format) or
standard 12-column BED
(https://genome.ucsc.edu/FAQ/FAQformat.html#format1)
format. It is recommended to use *short* and *unique*
sequence identifiers (such as Ensembl transcript id)
in FASTA and BED file. If this is a BED file,
reference genome ('-r/--ref') should be specified.
The input FASTA or BED file could be a regular text
file or compressed file (*.gz, *.bz2) or accessible
URL (http://, https://, ftp://). URL file cannot be a
compressed file.
-o OUT_FILE, --outfile=OUT_FILE
The prefix of output files.
-d LOGIT_MODEL, --logitModel=LOGIT_MODEL
Logistic regression model. The prebuilt models
for Human, Mouse, Fly, Zebrafish are availablel.
Run 'make_logitModel.py' to build logistic
regression model for your own training datset.
-x HEXAMER_DAT, --hex=HEXAMER_DAT
The hexamer frequency table. The
prebuilt tables for Human, Mouse, Fly, Zebrafish
are availablel. Run 'make_hexamer_tab.py' to make this
table for your own training dataset.
-r REF_GENOME, --ref=REF_GENOME
Reference genome sequences in FASTA format.
Reference genome file will be indexed automatically
if the index file ( *.fai) does not exist. Will be
ignored if FASTA file was provided to '-g/--gene'.
--antisense Logical to determine whether to search for ORFs
from the anti-sense strand. *Sense strand* (or coding
strand) is DNA strand that carries the translatable
code in the 5′ to 3′ direction. default=False (i.e.
only search for ORFs from the sense strand)
--start=START_CODONS Start codon (use 'T' instead of 'U') used to
define the start of open reading frame (ORF).
default=ATG
--stop=STOP_CODONS Stop codon (use 'T' instead of 'U') used to
define the end of open reading frame (ORF). Multiple
stop codons are separated by ','. default=TAG, TAA,
TGA
--min-orf=MIN_ORF_LEN
Minimum ORF length in nucleotides.
default=75
--top-orf=N_TOP_ORF Number of ORF candidates reported. RNAs may
have dozens of putative ORFs, in most cases, the real
ORF is ranked (by size) in the top several. It is not
necessary to calculate "Fickett score",
"Hexamer score" and "coding probability" for every
ORF. default=5
--width=LINE_WIDTH Line width of output ORFs in FASTA format.
default=100
--log-file=LOG_FILE Name of log file. default="CPAT_run_info.log"
--best-orf=MODE Criteria to select the best ORF: "l"=length,
selection according to the "ORF length";
"p"=probability, selection according to the
"coding probability". default="p"
--verbose Logical to determine if detailed running
information is printed to screen.
Examples
Use local FASTA file as input
$ cpat -x Human_Hexamer.tsv --antisense -d Human_logitModel.RData --top-orf=5 -g Human_test_coding_mRNA.fa -o output1
Use a remote FASTA file as input
$ cpat -x Human_Hexamer.tsv --antisense -d Human_logitModel.RData --top-orf=5 -g https://data.cyverse.org/dav-anon/iplant/home/liguow/CPAT/Human_test_coding_mRNA.fa -o output2
Use BED file as input. ‘-r’ is required
$ cpat -x Human_Hexamer.tsv --antisense -d Human_logitModel.RData --top-orf=5 -g Human_test_coding_mRNA_hg19.bed -r hg19.fa -o output3
Output
- 1. output.ORF_seqs.fa
The top ORF sequences (at least 75 nucleotides long) in FASTA format.
- 2. output.ORF_prob.tsv
ORF information (strand, frame, start, end, size, Fickett TESTCODE score, Hexamer score) and coding probability)
- 3. output.ORF_prob.best.tsv
The information of the best ORF. This file is a subset of “output.ORF_prob.tsv”
- 4. output.no_ORF.txt
Sequence IDs or BED entries with no ORF found. Should be considered as non-coding.
- 5. output1.r
Rscript file.
- 6. CPAT_run_info.log
The log file.
Build your own hexamer table
make_hexamer_tab calculates the in frame hexamer (6mer) frequency from CDS sequence in fasta format. A CDS is an mRNA sequence without the 3’ UTR and 5’ UTR regions.
This table is required by cpat to calculate the hexamer usage score. Users can download prebuilt hexamer tables (Human, Mouse, Fly, Zebrafish) from here.
Usage
$ make_hexamer_tab -h
make_hexamer_tab [options]
Options:
--version show program's version number and exit
-h, --help show this help message and exit
-c CODING_FILE, --cod=CODING_FILE
Coding sequence (must be CDS without UTR, i.e.
from start coden to stop coden) in fasta format
-n NONCODING_FILE, --noncod=NONCODING_FILE
Noncoding sequences in fasta format
Example
First, download these two files:
Coding CDS sequences: Human_coding_transcripts_CDS.fa.gz
Noncoding sequences: Human_noncoding_transcripts_RNA.fa.gz
Then, run:
$ make_hexamer_tab -c Human_coding_transcripts_CDS.fa.gz -n Human_noncoding_transcripts_RNA.fa.gz >Human_Hexamer.tsv
Output
$ head Human_Hexamer.tsv
hexamer coding noncoding
AAAAAA 0.0006471106736092786 0.001606589931772997
AAAAAC 0.00042092373222007566 0.0005113004850646316
AAAAAG 0.0008133623112408557 0.0006870944872085282
AAAAAT 0.0005917287586530271 0.0009504638599970318
AAAACA 0.0004934602747535982 0.0007256901384894673
AAAACC 0.0004003805362324795 0.0003686803641407804
AAAACG 9.064420497619743e-05 0.00010448394168197091
AAAACT 0.0004068399947646618 0.0004784022870680216
AAAAGA 0.0004286539039061299 0.000774026596998453
...
Build your own logit model
Build logistic regression model (“prefix.logit.RData”) required by cpat. This program will output
3 files:
prefix.feature.xls: A table contains features calculated from training datasets (coding and noncoding gene lists).
prefix.logit.RData: logit model required by CPAT (if R was installed).
prefix.make_logitModel.r: R script to build the above logit model.
Note: Users can download prebuilt logit models for Human, Mouse, Fly and Zebrafish.
Usage
make_logitModel [options]
Options:
--version show program's version number and exit
-h, --help show this help message and exit
-c CODING_FILE, --cgene=CODING_FILE
Genomic sequnences of protein-coding RNAs in
FASTA or standard 12-column BED format. It is
recommended to use *short* and *unique* sequence
identifiers (such as Ensembl transcript id) in
FASTA and BED file. The input FASTA or BED file
could be a regular text file or compressed file
(*.gz, *.bz2) or accessible URL (http://, https://,
ftp://). When BED file is provided, use the ORF
defined in the BED file (the 7th and 8th columns in
BED file define the positions of 'start codon,
and 'stop codon', respectively). When FASTA file
is provided, searching for the longet ORF. For
well annotated genome, we recommend using BED file
as input because the longest ORF predicted from
RNA sequence might not be the real ORF. If this is
a BED file, reference genome ('-r/--ref') should be
specified.
-n NONCODING_FILE, --ngene=NONCODING_FILE
Genomic sequences of non-coding RNAs in FASTA or
standard 12-column BED format. It is recommended to
use *short* and *unique* sequence identifiers (such as
Ensembl transcript id) in FASTA and BED file. The
input FASTA or BED file could be a regular text file
or compressed file (*.gz, *.bz2) or accessible URL
(http://, https://, ftp://). If this is a BED file,
reference genome ('-r/--ref') should be specified.
-o OUT_FILE, --outfile=OUT_FILE
The prefix of output files.
-x HEXAMER_DAT, --hex=HEXAMER_DAT
Hexamer frequency table. CPAT has prebuilt hexamer
frequency tables for Human, Mouse, Fly, Zebrafish. Run
'make_hexamer_tab.py' to generate this table.
-r REF_GENOME, --ref=REF_GENOME
Reference genome sequences in FASTA format.
Ignore this option if mRNA sequences file was provided
to '-g'. Reference genome file will be indexed
automatically if the index file *.fai) does not
exist.
-s START_CODONS, --start=START_CODONS
Start codon (use 'T' instead of 'U') used to
define the start of open reading frame (ORF).
default=ATG
-t STOP_CODONS, --stop=STOP_CODONS
Stop codon (use 'T' instead of 'U') used to
define the end of open reading frame (ORF).
Multiple stop codons are separated by ','.
default=TAG, TAA, TGA
--min-orf=MIN_ORF_LEN
Minimum ORF length in nucleotides.
default=30
--log-file=LOG_FILE Name of log file.
default="make_logitModel_run_info.log"
--verbose Logical to determine if detailed running
information is printed to screen.
Example-1: FASTA as input
make_logitModel -x Human_Hexamer.tsv -c Human_coding_transcripts_mRNA.fa.gz -n Human_noncoding_transcripts_RNA.fa.gz -o Human
2024-01-25 10:41:34 [INFO] Start codons used: "ATG"
2024-01-25 10:41:34 [INFO] Stop codons used: "TAG, TAA, TGA"
2024-01-25 10:41:34 [INFO] Reading hexamer frequency table file: "Human_Hexamer.tsv"
2024-01-25 10:41:34 [INFO] Process protein-coding RNA file: "Human_coding_transcripts_mRNA.fa.gz"
2024-01-25 10:41:34 [INFO] Protein-coding RNA file "Human_coding_transcripts_mRNA.fa.gz" is in FASTA format
2024-01-25 10:42:12 [INFO] Total 17984 coding sequences finished.
2024-01-25 10:42:12 [INFO] Process non-coding RNA file: "Human_noncoding_transcripts_RNA.fa.gz"
2024-01-25 10:42:12 [INFO] Non-coding RNA file "Human_noncoding_transcripts_RNA.fa.gz" is in FASTA format
2024-01-25 10:42:20 [INFO] Total 11519 non-coding sequences finished.
2024-01-25 10:42:20 [INFO] Wrting to "Human.feature.xls"
2024-01-25 10:42:20 [INFO] Making logistic regression model from "Human.feature.xls" ...
...
Example-2: BED as input
$ make_logitModel -x Human_Hexamer.tsv -c Human_coding_transcripts_hg19.bed -n Human_noncoding_transcripts_hg19.bed -r /database/hg19.fa -o Human
2024-01-25 10:44:45 [INFO] Start codons used: "ATG"
2024-01-25 10:44:45 [INFO] Stop codons used: "TAG, TAA, TGA"
2024-01-25 10:44:45 [INFO] Reading hexamer frequency table file: "Human_Hexamer.tsv"
2024-01-25 10:44:45 [INFO] Process protein-coding RNA file: "Human_coding_transcripts_hg19.bed"
2024-01-25 10:44:45 [INFO] Protein-coding RNA file "Human_coding_transcripts_hg19.bed" is in BED format
...
Use CPAT to detect ORF
When using CPAT to find ORFs, it will gives exactly the same results as NCBI ORFfinder.
Prepare data
Below is the mRNA sequence of protein-coding gene UQCR10. Copy and save it as “test.fa”.
>NM_013387.4
GCGGTGGCGCGAGTTGGACTGTGAAGAAACATGGCGGCCGCGACGTTGAC
TTCGAAATTGTACTCCCTGCTGTTCCGCAGGACCTCCACCTTCGCCCTCA
CCATCATCGTGGGCGTCATGTTCTTCGAGCGCGCCTTCGATCAAGGCGCG
GACGCTATCTACGACCACATCAACGAGGGGAAGCTGTGGAAACACATCAA
GCACAAGTATGAGAACAAGTAGTTCCTTGGAGGCCCCCATCCAGGCCAGA
AGGACCAGGTCCACCCAGCAGCTGTTTGCCCAGAGCTGGAGCCTCAGCTT
GAAGATGATGCTCAAGGTACTCTTCATGGACCACCATTCGCTGTTGGCAA
GAAACGGCTTTACTTACAAAACAGACTCTTTACCTTCTGCTGTGTTTGAA
GTATGTTTAGTCAGCATGCTCAGGAAATAAATGTGAATTGCCCTTGAGAC
CTGCTTCTACATTGGTTGCTTTGTTAACTCTACCTGATCTTCACTTGTCA
GTAATTTGAGACCACTTCAAAGCCCTCTGCAAACACCCCAAAGGCAGAAT
CTGCTATTTTGAGTTTTCCATTAACTTCCAAAGAATTCTGGTTTTCAAAA
CAGGAGCCAGAGTTGGAGATATTACAGTCAACTTTGGCTTCTAAGCCAGT
AATTCCATTCTTAAATACCTCACTGTCTTGGCCATGGGGAAGCACTATGG
CCTCAGCTGGGGGAAAGACCCTGGCCTAGGGGTCTTAGCCACTCCCCACC
CTAGGGTATAGTTCAGGGGTATCCAATCCTTTGGCTTCCCTGGGCCATGT
TGGAAGAATTGTCTTGGGCCACACATAAAATACAGTAACCATAGCTGATG
AGCTAAAACAAAAAACAATGGTTTGTGCAAAAATCTCATAATGTTTTAAT
AAAGTTGAAGAATTTG
Run CPAT
The command to run cpat.py is as below:
$ cpat -x Human_Hexamer.tsv -d Human_logitModel.RData --top-orf=100 --antisense -g test.fa -o output
Note
You must specify
--antisense, otherwise, it will only search ORFs from the sense strand.You also specify
--top-orfto a large number to report all the ORFs.The
--min-orfis set to 75 by default, same as NCBI ORFfinder.
Check the results
A total of 8 ORFs were found (sorted by the ORF size, the 7th column). If you copy and paste the same sequence to NCBI ORFfinder web server, you will get exactly the same results.
$cat output.ORF_prob.tsv
ID mRNA ORF_strand ORF_frame ORF_start ORF_end ORF Fickett Hexamer Coding_prob
NM_013387.4_ORF_1 916 - 2 327 1 327 1.103 0.28998918917275 0.792763525921043
NM_013387.4_ORF_2 916 + 2 209 430 222 1.1605 0.0674464550896935 0.271842476390681
NM_013387.4_ORF_3 916 - 1 889 695 195 0.9192 -0.32000518247443 0.0113140534730678
NM_013387.4_ORF_4 916 + 1 31 222 192 1.2952 0.600469985268255 0.915129459422605
NM_013387.4_ORF_5 916 - 1 337 197 141 1.1626 0.133867810597757 0.185245402415541
NM_013387.4_ORF_6 916 - 3 119 3 117 1.2673 0.442351820001225 0.618496534888714
NM_013387.4_ORF_7 916 - 3 842 735 108 0.5832 -0.19401829042094 0.00290794398512764
NM_013387.4_ORF_8 916 + 3 684 761 78 0.7415 -0.154613060436537 0.00454929869181486
CPAT offers Fickett’s TESTCODE score, Hexamer score, and coding probability for each ORF. While the largest ORF is often the most probable for most mRNAs, it’s not always the case. In the instance of NM_013387.4, the ORF_4 is the most likely to code for protein, evident from its highest coding probability, despite not being the largest ORF. This is confirmed through BLATing the 8 ORF sequences to the reference genome.

How to choose cutoff
Optimum cutoffs were determined from TG-ROC. For example, in human, coding probability (CP) cutoff >=0.364 indicates coding sequence, CP < 0.364 indicates noncoding sequence.
Species |
CP threshold |
Sensitivity & Specificity |
|---|---|---|
Human (Homo sapiens) |
0.364 |
0.966 |
Mouse (Mus musculus) |
0.44 |
0.955 |
Fly (Drosophila melanogaster) |
0.39 |
0.963 |
Zebrafish (Danio rerio) |
0.38 |
0.984 |
Here we provide the R code and the data that we used to generate Figure 3 in our paper. Note the ROCR library is required to run our R code.
1) Download R code and data from here
2) Put the R code and the data table in the same folder
$ ls
10Fold_CrossValidation.r Human_train.dat
3) Run the R code from command line or console. The R code will perform 10-fold cross validation and generate Figure_3.
$ Rscript 10Fold_CrossValidation.r # install ROCR before running this code
Loading required package: gplots
Attaching package: ‘gplots’
The following object is masked from ‘package:stats’:
lowess
Loading required package: methods
Warning message:
package ‘gplots’ was built under R version 3.1.2
[1] "ID" "mRNA" "ORF" "Fickett" "Hexamer" "Label"
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
null device
1
How to prepare training dataset
We prebuild hexamer tables and logit models for human, mouse, fly and zebrafish. If you want to run CPAT for other species, you need to prepare your own training data.
Optimal training datasets exhibit balance, where the count of coding sequences is approximately equal to that of noncoding sequences.
In cases where the genome of your target species lacks sufficient annotation of ‘coding’ and ‘noncoding’ genes for constructing a training dataset, consider leveraging data from evolutionarily related species to build your model.
Evaluating Performance
Figure-1
Combinatorial effects of 3 major features. 10,000 coding genes (red dots) and 10,000 noncoding genes (blue dots) are clearly separated into two clusters. (below figure)
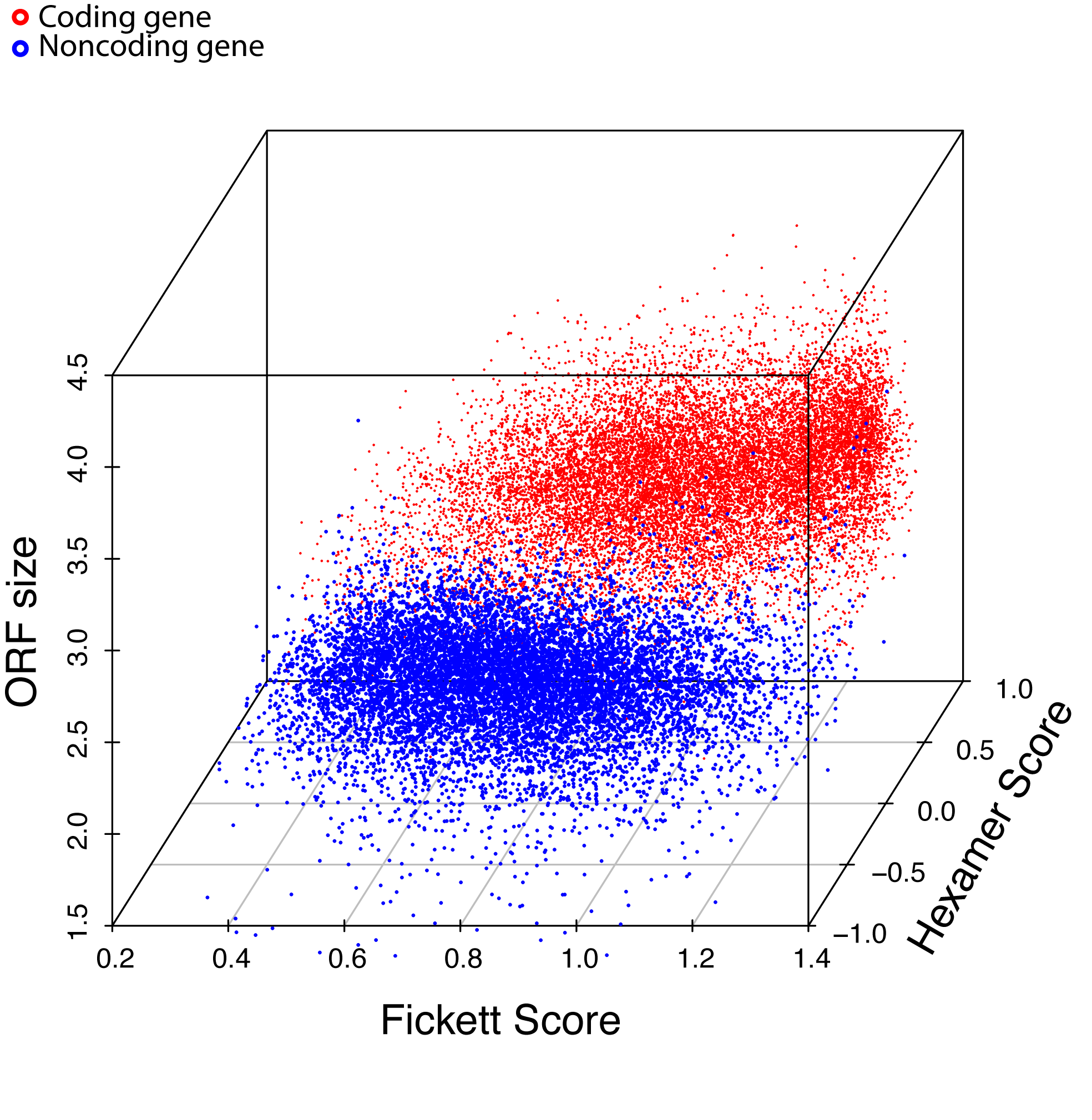
Figure-2
Performance evaluation was conducted using 10-fold cross-validation, considering a dataset comprising 10,000 coding genes and 10,000 noncoding genes. The blue dotted curves depict the results of individual 10-fold cross-validations, while the red solid curve represents the averaged curve across 10 validation runs. The evaluation metrics include: (A) ROC curve, (B) Precision-Recall (PR) curve, (C) Accuracy plotted against cutoff values, and (D) Two graphical ROC curves aimed at determining the optimum cutoff value.
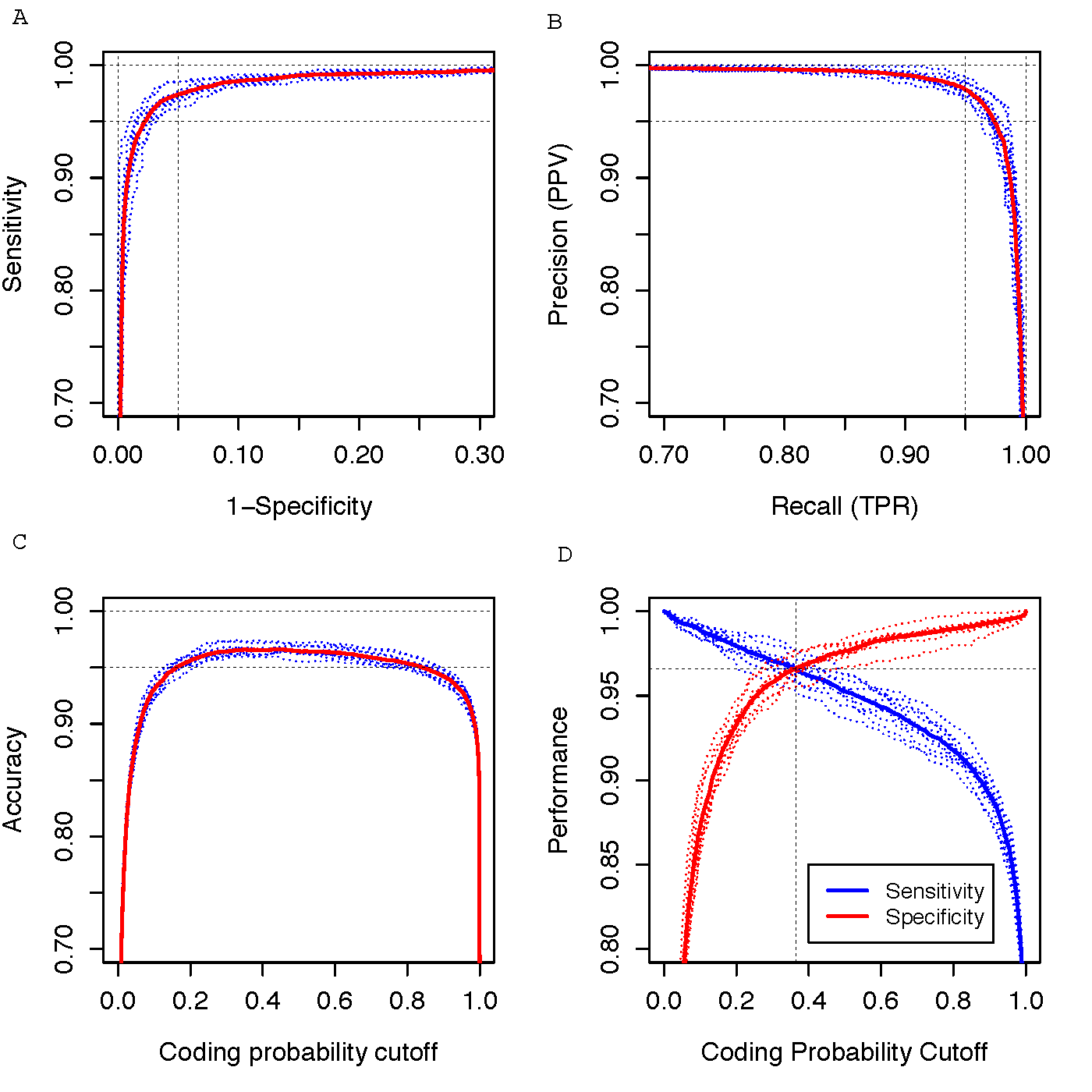
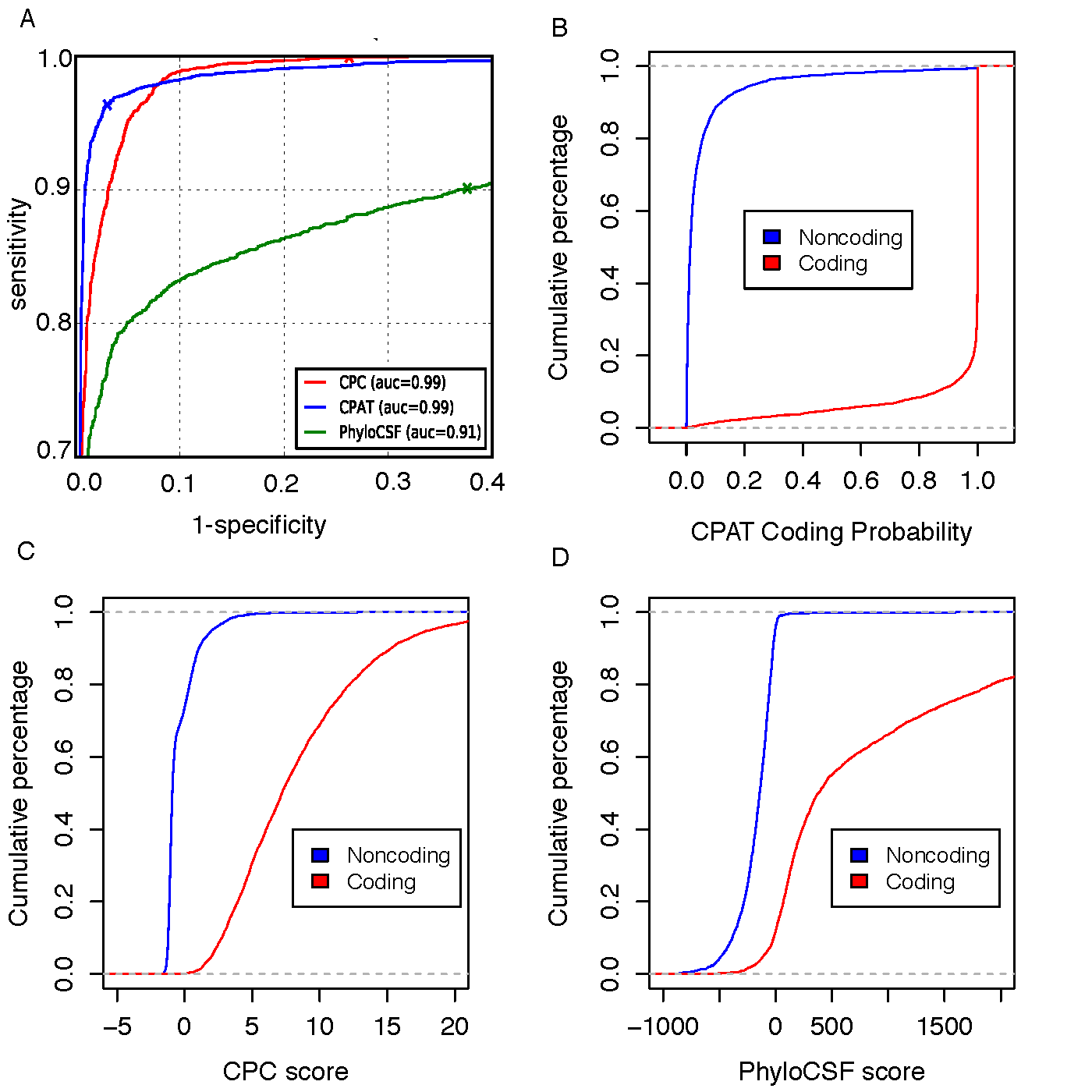
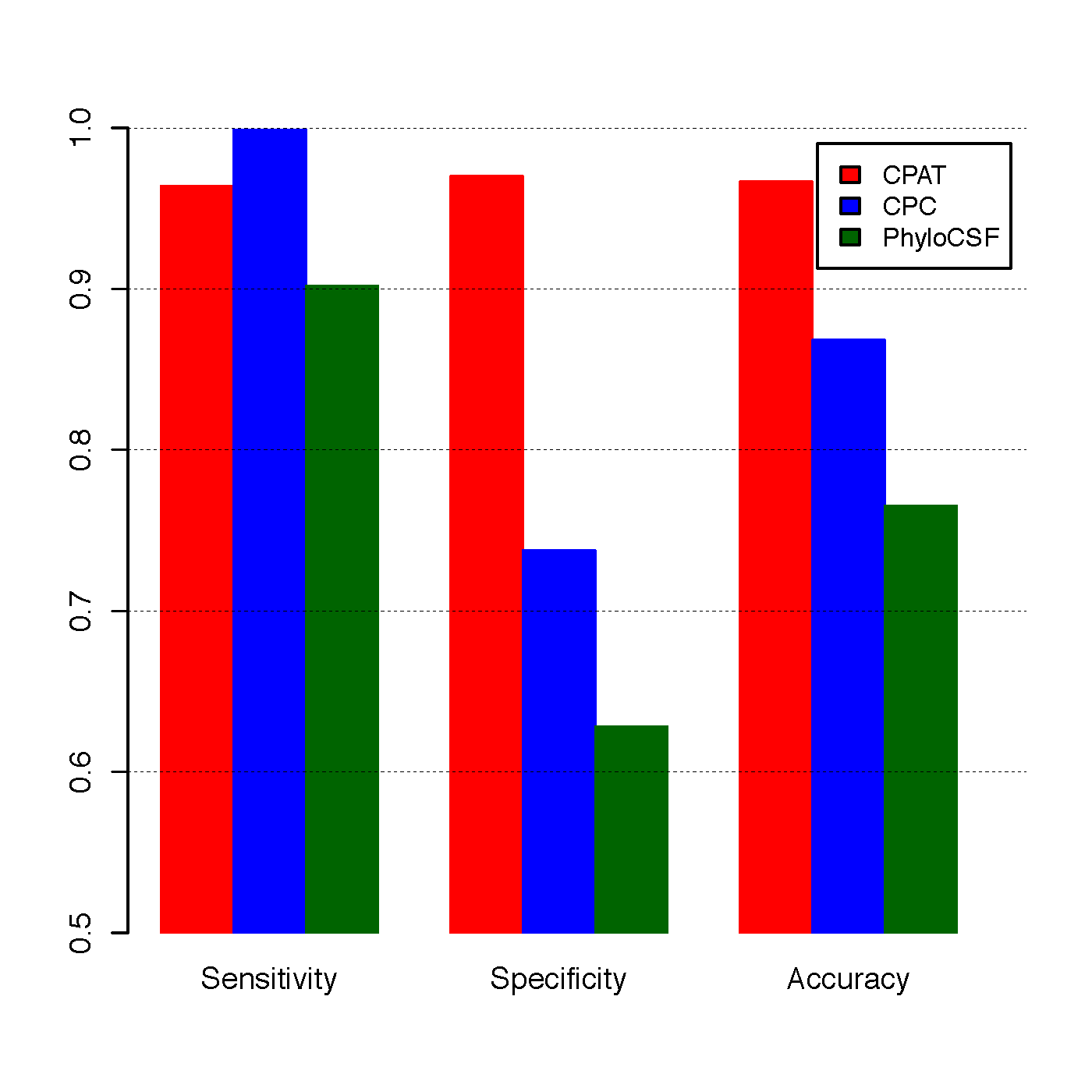
Figure-3
To compare CPAT with CPC and PhyloCSF, we build an independent testing dataset that composed of 4,000 high quality protein coding genes from Refseq annotation and 4,000 lincRNAs from Human lincRNA catalog (Cabili et al., 2011). All 8000 genes were not included in the training dataset of CPAT.
LICENSE
CPAT is distributed under GNU General Public License
This program is free software; you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation; either version 2 of the License, or (at your option) any later version. This program is distributed in the hope that it will be useful, but WITHOUT ANY WARRANTY; without even the implied warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU General Public License for more details. You should have received a copy of the GNU General Public License along with this program; if not, write to the Free Software Foundation, Inc., 51 Franklin Street, Fifth Floor, Boston, MA 02110-1301 USA
Reference
Wang, L., Park, H. J., Dasari, S., Wang, S., Kocher, J.-P., & Li, W. (2013). CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Research, 41(6), e74. doi:10.1093/nar/gkt006
Contact
Liguo Wang: wang.liguo AT mayo.edu
Wei Li: wei.li AT uci.edu